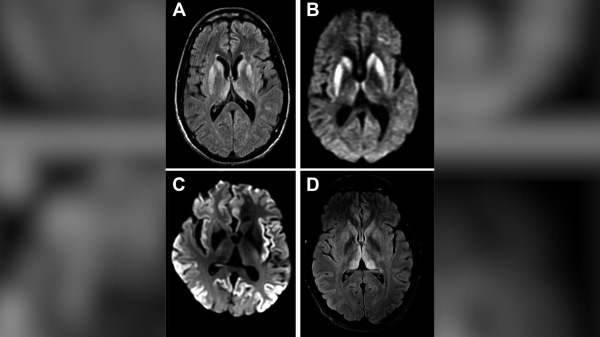
Магнітно-резонансна томографія (МРТ) сканування мозку пацієнта з ХКЯ. (Автор зображення: Pract Neurol, Mead S, Rudge PCJD mimics and chameleons Practical Neurology 2017; 17:113-121, CC BY 4.0, https://creativecommons.org/licenses/by/4.0/deed.en, через Wikimedia Commons, зображення на розмитому тлі)
Назва хвороби: хвороба Крейтцфельда-Якоба (ХКЯ), названа на честь Ганса Кройцфельда та Альфонса Якоба, двох німецьких лікарів, які вперше описали хворобу в 1920-х роках.
Уражене населення: CJD щороку вражає приблизно 1 з мільйона людей у всьому світі. У США щорічно діагностується приблизно 350 випадків ХКЯ. Чоловіки і жінки однаково схильні до розвитку захворювання.
Причини: CJD спричинений аномальними білками в мозку, відомими як «пріони». Ці пріони викликають незворотні пошкодження тканин, що призводить до утворення губкоподібних отворів у всьому мозку, які спричиняють неправильну роботу нейронів і їх загибель. Пріони викликають ланцюгову реакцію, спонукаючи інші, нормальні білки в мозку до неправильного згортання. Це закріплює стан і призводить до того, що пацієнти відчувають дедалі гірші проблеми з рухом і розумовими функціями.
Існує три основних типи CJD, які відрізняються залежно від того, як пріони утворюються в мозку. Найпоширенішою з цих форм є «спорадичний ХКЯ», на який припадає близько 85% випадків. Спорадичний CJD виникає, коли нормальні білки спонтанно неправильно згортаються і стають пріонами з невідомих причин, при цьому симптоми зазвичай спочатку розвиваються у дорослих віком від 45 до 75 років.
Крім того, від 10% до 15% випадків CJD викликані мутацією в гені під назвою PRNP, що призводить до розвитку пріонів. Ця генетична форма CJD успадковується аутосомно-домінантним типом, що означає, що дитині потрібно успадкувати лише одну копію дефектного гена від будь-якого з батьків, щоб розвинути захворювання. Генетичний CJD найчастіше зустрічається у людей у віці від 30 до 50 років.
Менше 1% випадків CJD є «інфекційними», тобто вони викликані передачею пріонів із зовнішніх джерел. Один із способів цього може статися, коли люди їдять яловичину від корів, які хворіють на губчасту енцефалопатію великої рогатої худоби, більш відому як «коров’яче сказство». З 1990-х років у США діють суворі правила, щоб цього не сталося. З моменту відкриття в 1996 році, що люди можуть отримати CJD від «скажених корів», у всьому світі було зареєстровано лише 233 таких випадки.
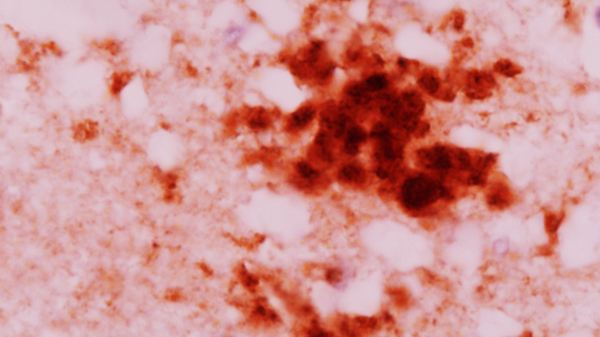
Пріони (коричневого кольору) можна побачити згрупованими разом у тканині мозку пацієнта з ХКЯ.
Ще один спосіб передачі CJD — випадкова передача від людини до людини під час медичних процедур — наприклад, якщо пацієнт отримує трансплантат або переливає кров від донора з CJD. Яскравий приклад цього стався з кінця 1950-х до 1985 року, коли лікарі давали пацієнтам заражені гормони росту, отримані з трупів. Це призвело до принаймні 226 випадків CJD у всьому світі, включаючи 29 випадків у США
Симптоми: поширеними симптомами CJD є деменція, сплутаність свідомості та дезорієнтація, галюцинації, відсутність координації та ригідність м’язів. У пацієнтів також можуть спостерігатися зміни особистості, сонливість, судоми та проблеми з розмовою. Крім того, вони можуть мати психологічні симптоми, такі як важка депресія, тривога та дратівливість.
Симптоми CJD часто швидко прогресують до точки, коли пацієнти стають повністю прикутими до ліжка, не помічають свого оточення та не можуть спілкуватися з оточуючими. CJD завжди є летальним, і приблизно 70% пацієнтів помирають протягом року після встановлення діагнозу, найчастіше через інфекцію, серцеву або легеневу недостатність.
Лікування: не існує ліків від CJD, але ліки можуть допомогти полегшити симптоми пацієнта. Наприклад, пацієнтам можуть призначати ліки для зменшення м’язових посмикувань або для полегшення тривоги.
Рання діагностика генетичної форми ХКЯ може допомогти пацієнтам, дозволяючи їм вжити заходів для догляду в кінці життя та планування сім’ї.
Останні випадки: у квітні 2025 року офіційні особи охорони здоров’я повідомили про три випадки ХКЯ, виявлені в осіб в окрузі Худ-Рівер в Орегоні протягом останніх восьми місяців — двоє з яких згодом померли. Поки ніякого зв'язку між випадками не встановлено.
Примітка редактора: цю статтю було оновлено 16 квітня 2025 року, щоб включити інформацію про нещодавні випадки в Орегоні. Стаття була вперше опублікована 13 лютого 2025 року.
Відмова від відповідальності
Ця стаття призначена лише для інформаційних цілей і не призначена для надання медичних порад.
ТЕМИ рідкісні захворювання

Emily CookeСоціальні посилання NavigationStaff Writer
Емілі – автор новин про здоров’я, живе в Лондоні, Велика Британія. Вона має ступінь бакалавра з біології в Даремському університеті та ступінь магістра з клінічної та терапевтичної неврології в Оксфордському університеті. Вона працювала в сфері наукових комунікацій, писала медичні статті та була репортером місцевих новин, одночасно проходячи навчання з журналістики NCTJ у News Associates. У 2018 році її назвали одним із 30 журналістів MHP Communications, за якими варто стежити молодше 30 років. (emily.cooke@futurenet.com)
Перш ніж коментувати, потрібно підтвердити своє загальнодоступне відображуване ім’я
Будь ласка, вийдіть, а потім увійдіть знову, після чого вам буде запропоновано ввести ваше відображуване ім’я.
Вийти
Sourse: www.livescience.com